

The barn door is on its last legs. Creaky hinges, rusty lock, maggot-infested wood, you name it. Thunder spooks the horses, and they rush out into the rain, trampling the door in their path. The farmer is worried his livestock won’t make it out in the open; he knows he need to corral them before they gallop too far away. So, he grabs his hammer and nails and starts reinforcing the beams of the door that gave way. Pause. That won’t work. Just as fixing a broken door won’t bring back horses that have already escaped, fixing problems in the cell that result in cancer after they have done their harm won’t cure the cancer either. If cancerous molecules have already been released into the body, why spend time repairing the mutation that produced them?
At the Yale Cancer Center, Dr. Peter Glazer and Dr. Ranjit Bindra TC ’98 attacked this problem. Cellular mutations that disrupt two genes called IDH1 and IDH2 break the barn door, so to speak, and release a tumor-causing molecule called 2HG. Glazer and Bindra found that cells with these mutations were more susceptible to an existing class of drugs called PARP inhibitors. Instead of attempting to repair the door and reverse the mutation, as many current therapies do, PARP inhibitors exploit this cellular weakness by preventing DNA repair. Beyond establishing a new link between IDH mutations and DNA repair, the research paves the way for a novel, highly promising cancer therapy.

Broken brakes
Though it has a single name, cancer is no one disease. Rather, it refers to any time a population of cells in the body begins to divide uncontrollably and spread. This definition may seem counterintuitive. Obviously, we need our cells to divide, or else we’d still be single-celled; we need different populations of cells to migrate, or else we’d never fight off infection or heal wounds. Normal cells, however, know when and where to proliferate because the cell cycle, the process by which cells divide, is tightly regulated. The body makes new cells as needed and no more. When this mechanism goes awry, cells will continue to proliferate past the point of necessity and often form tumors.
A major switch that triggers this dysregulation of the cell cycle is mutation, or a change in a cell’s DNA sequence. Not just any mutation will result in cancer. Relevant genes are those already implicated in cell division. Genes that normally promote proliferation can be mutated into can be mutated to become overactive, causing cancer. IDH2 mutations fall into a second category, where mutations deactivate tumor-suppressing genes.
BRCA’s buddy
The tumor-causing 2HG molecules produced when the IDH genes are mutated are called oncometabolites. The prefix onco- means tumor, and metabolite means that it is involved with cellular processes. Taken together, an oncometabolite is an unintended product of cellular processes that can disrupt the cell cycle and drive tumor formation. 2HG is like smoke coming out of an engine—something had to go wrong in the car for it to be produced, and the smoke further pollutes the air. Most current approaches to treating cancer through this mechanism aim to target IDH1/2 mutations and stop the oncometabolite from being produced. Drugs that operate under this logic are currently in clinical trials and were widely accepted as the cutting edge of cancer research. However, when Glazer and Bindra worked together to tackle this same problem, they saw it from a completely new angle.
The two doctors’ relationship traces back to 2006, when Bindra was an MD/PhD student in Glazer’s lab. They studied DNA repair under hypoxia, in which cells are oxygen-deprived; hypoxic conditions frequently spur cancer formation. After completing his residency and fellowship at Memorial Sloane Kettering Hospital in New York, Bindra returned to New Haven in 2012 and was given his own lab in Glazer’s department. By 2013, Bindra had looked for specific genetic defects in tumors that could serve as targets for drug therapies, identifying the IDH1/2 mutations as prime targets. Computer models of the mutant genes showed that PARP inhibitor drugs could be used to distinguish between normal cells and those with IDH1/2 mutations. At this point, Glazer realized the potential of this project and rejoined forces with Bindra. “We realized that if this was true, it was probably going to change clinical practice,” Glazer said.
A common treatment for breast and ovarian cancer, PARP inhibitors target mutations in the BRCA1 and BRCA2 genes. Angelina Jolie’s famous disclosure that her maternally inherited copy of mutant BRCA1 was the reason for her double mastectomy piqued women’s interest in their own vulnerability. Both referrals to genetic testing facilities and questions about preventative surgery significantly increased in a phenomenon now termed the “Angelina Jolie effect.” BRCA1/2 mutations render cells incapable of putting their DNA back together, which made them susceptible to carcinogenic mutations. Instead of reversing the mutation’s effects, PARP inhibitors further unstitch the DNA to the point where tumor cells simply fall apart. When Glazer and Bindra found that IDH1/2 mutations were associated with PARP inhibition, they reasoned that these genes could do something similar to BRCA1/2.
Back to basics
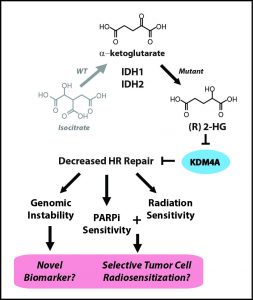
To identify potential targets for PARP inhibiton, the researchers developed the criterion “BRCAness,” or similarity of a given gene to the BRCA1/2 genes. A mutation with high BRCAness is heavily implicated in deficient DNA repair and, thus, a strong candidate target for PARP inhibitors. Bindra and Glazer knew that IDH1/2 mutants produced the oncometabolite 2HG, but it was still unclear how the gene mutations blocked DNA repair. To isolate the mutation, the team turned to classical genetics, the basic patterns of gene inheritance discovered by Gregor Mendel. Past IDH1/2 research had not been very selective in choosing tumor cell lines, using any cells that happened to contain the relevant mutations. They chose not to have a delicately engineered model for two reasons: first, this approach is quick—one can screen for the mutation and, if it is present, immediately begin experimenting. Second, a human tumor cell line with a naturally occurring IDH1/2 mutation is more relevant to actual cancer patients than one with a more refined genome. Yet Bindra and Glazer took the time to cross cells over the course of generations and grow a proper single gene mutation culture of cells. As a result, they were able to study its specific effects without worrying about confounding variables.
Once they established this solid foundation, the rest fell right into place. The results they observed were so robust that even at a glance, the statistical significance was clear. The first string of results strongly suggested that cells with the IDH1/2 mutation cannot repair breaks in DNA and are targeted by PARP inhibitors. Next, the team demonstrated that 2HG was responsible for both these properties. “It created an unsuspected vulnerability, like an Achilles heel,” Glazer said. Even though the unchecked result of IDH1/2 mutations is cancer, 2HG’s susceptibility to PARP inhibitors is a chink in the armor that gives medicine a chance to intervene. Now that the team had established baseline results in their classically crafted cell line, they repeated their tests in samples of brain tumor cells, which yielded similar results. Finally, they administered an FDA-approved PARP inhibitor called olparib to mice with the IDH1 mutation, and saw cell death in brain tumors increase 50-fold. Considering that unchecked cell proliferation causes cancer, this striking halt in tumor growth holds great promise for the future of cancer treatment.
Beneficial breaks
When Glazer and Bindra first published their findings, they were met with resistance. Reviewers couldn’t fathom how other scientists had glossed what appeared to be a conceptually straightforward idea and so were hesitant to publish findings that so directly undermined the norm in this branch of cancer drugs. Once the story was out, however, it quickly received major recognition. The team has received calls from PARP inhibitor companies, patient advocates, and patient support groups, among others. “The story wasn’t just kicking the can down the road,” Bindra said. He reflected that the team’s achievement is a prime example of why the NIH funds academic science—without the leeway to truly delve into the biology, such progress would not have occurred.
Moving forward, Glazer and Bindra have scheduled clinical trials for olparib, which is currently only used on ovarian cancer patients, in 35 different medical centers nationwide. They hope to expand its use to brain cancer, liver cancer, leukemia, and more. There is also potential for 2HG to be used as a biomarker for a cancer patient’s sensitivity to PARP inhibitors. Since 2HG is not specific to one type of tumor, its use as a biomarker can be extended to many different types of cancer. Both these applications are the result of a major breakthrough in our understanding of how cancer develops. “You spend a lot of time in a lab either talking to biology or listening to it,” said Bindra. “This is one of those moments where you hit something and biology just started talking like crazy.”
Featured Image: While current therapies aim to remedy the IDH mutations and so stop production of oncometabolites, these carcinogenic molecules have already been released into the body. The research team reasoned that fixing the barn door is useless once the horse has escaped.
About the Author:
Charlie Musoff is a freshman in Davenport College and a prospective molecular, cellular, and developmental biology major. Besides being Yale Scientific’s Outreach Designer, Charlie enjoys running with Yale Club Running, singing with the Baker’s Dozen, and teaching with Community Health Educators.
The author would like to thank Drs. Peter Glazer and Ranjit Bindra for their time and insights.
Further Reading: Corso, Christopher D., MD PhD, and Ranjit S. Bindra, MD PhD. “Success and Failures of Combined Modalities in Glioblastoma Multiforme: Old Problems and New Directions.” Seminars in Radiation Oncology 26 (2016): 281-98. Science Direct. Web.