
Image generated using ChemDraw.
Nucleotides, the building blocks of DNA and RNA, can be synthetically designed to mimic their biological counterparts and interact with the cellular environment. An important class of nucleotide molecules are cyclic dinucleotides (CDNs), two single nucleotides that bind to form a ring. For example, cGAMP is a naturally occurring CDN that can drive immune response by activating the stimulator of interferon genes (STING) protein. Because interferon genes are critically involved in immune cell signaling, STING pathways are being extensively evaluated in cancer immunotherapies, and CDNs such as cGAMP may serve as promising new drugs. Synthetically manufacturing them represents an important step in bringing these drugs to mass market.
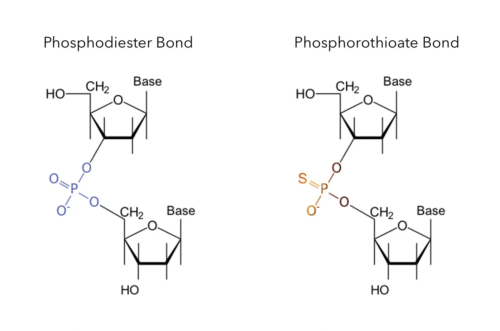
A powerful method of increasing metabolic stability and therapeutic potential of CDNs is to modify their native phosphodiester linkages into phosphorothioate (PS) double-bonds. However, doing so introduces an additional layer of complexity by rendering the central phosphorus (P) atoms stereogenic (chiral), meaning different spatial orientations—stereoisomers—of the same connected groups are possible. An analogy for stereoisomers is a pair of baseball gloves in which one is left-handed and the other is right-handed—although otherwise indistinguishable, the right-handed glove will not fit on a left hand, and vice versa.
Each CDN stereoisomer, despite possessing identical atomic connectivities, displays markedly different physical properties and biological activities. The importance of stereochemistry is best highlighted by the infamous case of thalidomide, a drug developed in the 1950s that was marketed to treat morning sickness in pregnant women. One of its stereoisomers has sedative effects, whereas the other caused birth defects. As a result, thousands of children whose mothers took thalidomide during pregnancy had severe birth defects, since both stereoisomers were present in the medicine.
Most synthetic methods for preparing PS-derived CDNs are nonselective in stereochemical outcome, relying on intensive purification techniques or large quantities of auxiliary reagents to produce stereopure products. In a recent study published in Science, the Scott Miller lab at Yale’s Department of Chemistry, in collaboration with Takeda Pharmaceutical Company, reported the first catalytic, stereocontrolled synthesis of dinucleotides. Lead author Aaron Featherston, along with a team of academic and industrial researchers, sought to address the aforementioned problem using asymmetric catalysis. Catalysts, which are regenerated throughout the course of a reaction, could ideally be used in small amounts to facilitate the stereoselective assembly of PS compounds.
The first step of oligonucleotide synthesis is often achieved using excess amounts of an acid activator. Symmetric chiral phosphoric acids (CPAs) have proven to be powerful catalysts in several asymmetric transformations that isolate one stereoisomer. Recently, the Miller lab developed an additional class of CPAs containing a modified amino acid, phosphothreonine (pThr). Their rationale was that the amino acid, a building block of proteins, could conceivably help impart selectivity on par with enzymes, the ultra-specific protein catalysts of nature.
Accordingly, Featherston and the team assessed the ability of these two classes of CPAs to asymmetrically couple two select nucleotides, guanosine and adenosine. They found that several pThr-derived catalysts greatly biased coupling towards one product stereoisomer. Alternatively, the symmetric CPA catalysts were highly selective for the other stereoisomer. The researchers thus demonstrated two distinct, complementary catalyst frameworks, allowing for the synthesis of either stereopure isomer.
The researchers then evaluated the ability of these catalysts to asymmetrically couple a broader range of representative nucleotides, finding that their stereoselective catalyses could be applied to other base pairs beyond the guanosine-adenosine coupling initially tested. Although selectivity decreased for certain nucleotide couplings, the chemists demonstrated that their approach is generalizable and can potentially yield a universal catalyst. Finally, the group applied their method to the synthesis of PS-modified cGAMP, showing that the pThr and symmetric CPA catalysts could allow for an efficient, stereocontrolled preparation.
The researchers found, for the first time, a stereocontrolled synthesis of dinucleotides using two catalyst frameworks. Oligonucleotide syntheses have long struggled with controlling P-stereogenicity, which reduces the efficacy of nucleotide-based therapeutics. This new approach both provides efficient access to a dinucleotide P-stereoisomer and enables preparation of stereopure analogs of cGAMP, a CDN of great biological significance.
In the future, the Miller lab may dive deeper into the catalysts’ mechanism of action. A longer-term ambition would be to develop a more universal catalyst framework that can facilitate the asymmetric synthesis of all possible nucleotide couplings. Their current findings will prove vital to the field of oligonucleotide chemistry, as synthetically modified nucleotides continue to grow in importance. “It was a great collaboration, and I think we were able to make a significant impact by developing some really cool chemistry,” Featherston said.
Citations:
Featherston, A. L., Kwon, Y., Pompeo, M. M., Engl, O. D., Leahy, D. K., & Miller, S. J. (2021). Catalytic asymmetric and stereodivergent oligonucleotide synthesis. Science, 371(6530), 702–707.